Introduction:
As our
knowledge of protein-protein and protein-DNA interactions has been greatly
increased with the availability of a large number of cocrystal structures,
protein-RNA interactions remain hard to predict even for interactions in which
only a single amino acid mutation has been introduced. With protein-protein and
protein-DNA interactions, the large amount of incoming structural data is
beneficial to measuring the accuracy of the current structural prediction tools,
yet tools that use protein-RNA interactions to predict structure still require
more development before accurate structural prediction can be achieved.
Understanding protein-RNA interactions is essential to predicting structures for
large complex molecules that contain both protein-protein interactions and
protein-RNA interactions. This study focuses on a mutation (Phe56Ala) of a
particular protein-RNA interaction between the N-terminal RNP domain of the U1A
RNA binding protein and stem loop 2 of U1 snRNA, which is just one of the many
interactions occurring within a larger molecule the spliceosome. [The
spliceosome is an important collection of molecules designed to cut out (splice)
introns (non-coding pieces of RNA) from transcribed RNA, leaving only the exons
(coding pieces of RNA) to be translated into protein by the ribosome.]
The Phe56Ala mutation results in a five order of magnitude decrease in
binding affinity for the interaction, which is a significant change due to a
single amino acid mutation. By comparing the mutant structural and thermodynamic
data with that of the wild type, we can study specific interactions that will
help to predict structural changes in protein-RNA complexes, as well as the
changes in thermodynamics that accompany the structural changes. Of course, the
result of this study alone will not solve all protein-RNA structures, but the
valuable in depth study of this particular interaction (rather than attempting
to extract patterns from a database of information) will help to answer one of
the most important structural biology questions to date: namely how can the
tertiary and quaternary structure of a protein be predicted from primary
structure? Answering this question could provide invaluable information in
nearly every scientific field, especially comparative genomics and rational drug
design.
Progress Report:
In order to perform X-ray analysis, of course, purified protein must be
crystallized. However, obtaining
pure U1A RNA binding protein can be equally complicated as the crystallization
itself. The original clones of U1A
protein produced product that contained minor contaminant RNA nuclease activity.
Since the project involves crystallizing protein/RNA complexes, RNAse
must be separated from the protein before crystallization can be performed.
In order to separate the U1A protein from RNAse, our goal is to engineer
a N-terminal poly(HIS) to leader segment, which should bind to a Ni2+
chelated purification column, providing an efficient means of separation.
The U1A protein has proven extremely difficult to transform into the
pET14b plasmid; a step necessary for controlled overexpression of the protein in
E. coli. Starting with a plasmid
containing wild type U1A sequence (provided from Nagai’s lab), I designed a
pair of PCR primers to extract and amplify a 92 amino acid section of the 97
amino acid U1A sequence. According
to crystallization experiments performed by Nagai on the wild type U1A protein,
the deletion of the five amino acids near the N-terminus of the protein afforded
the highest resolution crystal without sacrificing structural accuracy of the
protein1. The goal of the
PCR procedure was to isolate the U1A sequence from the Nagai plasmid so that the
sequence could be transformed into a plasmid with controlled induction to aid in
producing a large amount of U1A protein. Despite
obtaining a fairly pure PCR product (Figure 1.)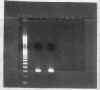 , the ligation of the PCR insert into the
pET14b plasmid proved to be very inefficient.
Most of the ligations transformed very well into the XL-1 Blue strain of
E. coli, but when screened with a restriction digest, none of hundreds of
colonies (from tens of ligations) appeared to contain the U1A insert.
, the ligation of the PCR insert into the
pET14b plasmid proved to be very inefficient.
Most of the ligations transformed very well into the XL-1 Blue strain of
E. coli, but when screened with a restriction digest, none of hundreds of
colonies (from tens of ligations) appeared to contain the U1A insert.
Success
was achieved after sequencing the only colony screened that contained the ~300
base pair U1A insert (though the presence of other bands of DNA in the screening
initially indicated that we did not have the right insert, or that the insert
had been mutated in some fashion). Sequencing
data of the questionable plasmid revealed that three U1A inserts had actually
ligated into the pET14b plasmid in some fashion; all in the same direction.(Figure
2.)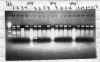 Since the inserts were all in the same direction back to back on the
plasmid, the final step in obtaining the plasmid that we wanted involved cutting
out two of the U1A inserts with restriction enzymes (BamHI, StuI, or NdeI).
BamHI was ruled out because the end of the first insert lacked a BamHI
due to a mutation of the sequence from C to G in the last base, thus, this
enzyme could never cut out two of the inserts.
StuI was rejected because DCM methylation interferes with the activity of
the enzyme, and although StuI site did not contain a DCM sequence, which would
have completely abolished StuI activity, there was a flanking DCM sequence only
two bases away from the beginning of the StuI site. Since
we had tried to cut our insert with StuI earlier and failed, we concluded that
the flanking DCM sites reduced the activity of StuI to a negligible amount.
Left with NdeI as our only option, we cut the pET14b plasmid with NdeI
and obtained promising results. We
are in the process of screening colonies produced from a ligation of the cut
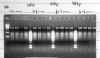
NdeI plasmid, and the most recent screening indicates a functional plasmid.(Figure
3.)
Since the inserts were all in the same direction back to back on the
plasmid, the final step in obtaining the plasmid that we wanted involved cutting
out two of the U1A inserts with restriction enzymes (BamHI, StuI, or NdeI).
BamHI was ruled out because the end of the first insert lacked a BamHI
due to a mutation of the sequence from C to G in the last base, thus, this
enzyme could never cut out two of the inserts.
StuI was rejected because DCM methylation interferes with the activity of
the enzyme, and although StuI site did not contain a DCM sequence, which would
have completely abolished StuI activity, there was a flanking DCM sequence only
two bases away from the beginning of the StuI site. Since
we had tried to cut our insert with StuI earlier and failed, we concluded that
the flanking DCM sites reduced the activity of StuI to a negligible amount.
Left with NdeI as our only option, we cut the pET14b plasmid with NdeI
and obtained promising results. We
are in the process of screening colonies produced from a ligation of the cut
NdeI plasmid, and the most recent screening indicates a functional plasmid.(Figure
3.)
Goals for the Academic Year:
With the plasmid needed to produce U1A protein that can be purified
nearly constructed, I am now confident that I will be able to purify protein and
obtain the preliminary X-ray crystal data by the end of the year.
First, I must perform two mutagenesis reactions on the U1A insert.
In addition to the F56A mutation, two other mutations (Y31H and Q36R) are
required to obtain high resolution crystals of U1A protein1. I
do not expect the difficulties of transforming the PCR insert to arise with the
mutagenesis inserts because mutagenesis is a notably much less sensitive
procedure, and obtaining the final plasmid for overexpression should take a few
weeks. The U1A protein has been
HIS-tagged for easy (in a relative sense) purification, which should not take
more than a few weeks. Assuming that
the mutated protein crystallizes under the same conditions and to the same
resolution (or at least a reasonable resolution for structure determination) as
the wild type protein1, crystallization analysis should be complete
within six months of obtaining pure protein.
Plan for the Academic Year:
1.
Experiments
The majority of the work to obtain the “ingredients” of this project
results from molecular biology experiments designed to purify U1A protein in an
efficient way so that X-ray diffraction data may be gathered for analysis by
computer software. The pET14b
expression vector is a perfect tool for achieving this goal.
The pET14b plasmid contains a “HIS-tag,” a poly histidine encoding
region located in the multi-cloning site of the plasmid that adds or “tags”
a histidine region to the N-terminus of the protein that is being expressed.
The purpose of adding this tag to the protein is that when a his-tagged
protein is run over a Ni2+ chelated purification column, the protein
may be separated very efficiently from cellular protein extract as long as the
tag is not buried within the protein (we think that it is not).
A simple wash of the column will yield pure protein that is essential for
forming ordered protein crystals. In
addition to the histidine encoding region, pET14b has an inducible lac operon preceding the multi-cloning site that allows for
controlled expression of the gene inserted into the plasmid (the U1A insert).
The operon (induced by the molecule IPTG) is essential to overexpressing
the protein of interest and obtaining the highest yield of protein possible.
Since we are forcing the cell to make an amount of protein in drastically
high quantity, eventually, the cell will become unhappy and perish.
By controlling the on/off switch to protein production, we can harvest
the protein from cells at our leisure, producing a large amount of protein in a
relatively short amount of time with a manageable number of cells.
Once purified protein has been obtained, the protein must be crystallized
in order to perform X-ray diffraction analysis to determine the structure.
Protein crystallization in itself is a separate science.
There is very little known about prediction of crystallization conditions
of proteins like there are for most small molecules.
The basic idea is simple, if a solution of pure protein becomes
supersaturated, crystals will spontaneously form and grow.
However, the more supersaturated a protein becomes in solution, the
greater tendency that protein will have to create the “critical nuclei,”
which are the first aggregates of molecules that eventually form crystals.
The small microcrystals formed in solutions that are highly
supersaturated are useless for X-ray diffraction analysis because the generation
of interpretable diffraction data is dependent on the presence of many protein
molecules gathered together in a highly ordered fashion to amplify the
diffraction of the X-rays by each molecule.
Therefore, we do not want all of the protein to form microcrystals, but
large macrocrystals that contain large amounts of protein molecules.
The region where protein crystals grow but are not formed is defined as
the supersaturated metastabile region, which lies between the undersaturated
region, where crystals dissolve, and the supersaturated labile region, where
crystals form and grow. The
objective of protein crystallization is to bring the solution containing the
protein of interest (U1A) into the labile region for only a brief period
allowing crystals to initiate formation of critical nuclei, then adjusting
conditions such that the solution is maintained in the metastabile region
thereafter, so that the crystals will grow to a considerable size2.
Crystallographers usually start
out by testing a wide range of conditions that differ in a number of variables
including salt concentration, pH, temperature, or ionic strength unless they
have reason to believe that the protein of interest will crystallize in a
similar manor to another protein. Solutions
that show promising crystals are then reduced by finer and finer gradients until
the optimal crystallization conditions are achieved.
Sometimes the protein must be mutated or deleted in non-functional areas
in order to achieve suitable crystals, that is, crystals that diffract to a
resolution of around 3 angstroms or less (protein crystals rarely diffract to a
resolution of less than 1). The
process of obtaining a protein crystal can take months and even years to
complete. In our case, two mutations
as well as deletions at the N-terminus of the U1A protein were made to achieve a
suitable crystal by Nagai1. We
expect that the addition of the his-tag and the mutation to our Phe56Ala mutant
will not affect the crystallization protocol significantly, but if this
assumption is proven false, the his-tag may be cleaved from U1A by the enzyme
thrombin. The mutation is also
expected not to alter the overall structure of the U1A protein to a large
degree, thus, refinement techniques such as heavy atom analysis should not be
necessary on the experimental level. The
X-ray analysis of U1A will be performed at VCU at the Center for Structural
Biology and Drug Discovery.
2. Computation
Once the diffraction data for the
U1A protein has been obtained, an electron density map will be generated by a
Fourier synthesis based on the triple sum of the amplitude, reflection and phase
of the electrons in three dimensional space divided by the volume of the unit
cell (the length times width times height of the protein dimensions), which can
all be extrapolated from the diffraction data of the crystal (the pattern and
intensity of diffraction). The
program we will use to visualize this electron density map is X-tal View.
Initial electron density maps will be calculated from the X-ray
diffraction data collected from the mutated U1A crystal and phasing information
from the wild type U1A structure (which has already been determined).
Refinement of the phases will come from careful model manipulation of the
mutant protein. For the U1A Phe56Ala
mutant protein, my contribution involves taking the basic structure of the
native U1A protein, and adjusting this structure with X-tal View to fit the
electron density map generated from the X-ray diffraction data of mutant U1A.(Figure
4.)
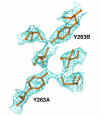 Eventually, the two U1A protein structures will be compared to describe
the effects of the Phe56Ala mutation on the structure of the protein.
A similar analysis will be performed with the mutated U1A/U1A RNA
complex.
Eventually, the two U1A protein structures will be compared to describe
the effects of the Phe56Ala mutation on the structure of the protein.
A similar analysis will be performed with the mutated U1A/U1A RNA
complex.
As an additional task, the
experimental protein structural will be compared to the minimized predicted
structure using HINT (Hydropathic INTeraction) software in conjunction with the
SYBYL program. The predicted
structure has already been determined by Peter Likarish of Dr. Kellogg’s (VCU)
lab using SYBYL. The interactions
between the U1A protein and its RNA substrate will be calculated by HINT.
This data will be compared to the experimental thermodynamic data
gathered by Dr. Baranger (
Wesleyan
University
) on the Phe56Ala mutant. Only by
comparing the predicted data of computer programs such as HINT and SYBYL will we
be able to refine the programs to more accurately predict the effects of
mutations on the structure of proteins, RNA and DNA, and the interactions
between them. Eventually the goal of
all structural biologists and chemists is to develop programs capable of
determining the accurate structure of all molecules, and the thermodynamics of
those interactions, making wet lab work unnecessary for structure determination.
3. Courses
I will not be taking any specific courses on X-ray crystallography during
the coming academic year, but I will be taking Biochemistry, which will enhance
my molecular biology knowledge as well as provide insight as to what may be the
U1A protein’s role in the spliceosome. I
will also be taking Physical Chemistry, which should help increase my basic
understanding of the physical principles behind crystallography, and how to
interpret the data obtained from the other commonly used chemical analysis
techniques.
Budget:
The budget for this project is as yet unknown, but will be funded by
grants from NIH and NSF, in cooperation with the BBSI program at VCU.
References:
1. Oubridge, Chris
et. al. "Crystallisation of RNA-Protein Complexes II. The Application of
Protein Engineering for Crystallisation of the U1A Protein-RNA Complex." J.
Mol. Biol. (1995) 249, 409-423.
2. McPherson,
Alexander. Crystallization of
Biological Macromolecules.
Cold Spring Harbor
,
New York
:
Cold
Spring
Harbor
Laboratory Press. 1999.
Pgs. 133 and 135.
3. Creighton,
Thomas F. Proteins: Structure and
Molecular Properties.
New York
: W. H. Freeman and Company. 1984.
Pgs. 212 and 213.
Acknowledgments:
I particularly want to thank Dr. Jeff Elhai for helping to organize the
entire BBSI undertaking as well as my VCU mentor Dr. Jason Rife, without whose
help and guidance with my repetitive questions, I would not have had the
knowledge to undertake this project. I
also want to thank Heather O’Farrell for guiding me through some of the tough
molecular biology enigmas as well as helping with some of the dirty work early
in the morning.